Bioequivalence Study of Dexrabeprazole Gastro Resistant Tablets in Healthy Male Subjects Under Fasting and Fed Conditions
G. Demiray1*, P. Özdüven2, E. Durucu3, M. Bilgiç4, H. Aydonat1, B. Güney1, E. Doğan-Kurtoğlu1, S. Bilgiç4, R.S. Alpan2, M. Nacak3, O. Sağlam1, A. Erenmemişoğlu5
1 Novagenix Bioanalytical Drug R&D Centre, Ankara, Turkey.
2 TNC Pharmaceuticals R&D Ltd. Istanbul, Turkey.
3 Farmagen-Good Clinical Practice Center, Gaziantep, Turkey.
4 Neutec İlaç San. Tic. A.Ş.-Turkey.
5 Alpan Farma R&D Biotechnologies Ltd., Istanbul, Turkey.
*Corresponding Author
Gökçe Demiray,
Novagenix Bioanalytical Drug R&D Centre, Ankara, Turkey
E-mail: gdemiray@novagenix.com
Received: November 04, 2022; Accepted: February 09, 2023; Published: February 20, 2023
Citation: G. Demiray, P. Özdüven, E. Durucu, M. Bilgiç, H. Aydonat, B. Güney, et al., Bioequivalence Study of Dexrabeprazole Gastro Resistant Tablets in Healthy Male Subjects Under Fasting and Fed Conditions. Int J Bioanal Methods Bioequival Stud. 2023;6(1):93-99.
Copyright: G. Demiray© 2023. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Rabeprazole is a racemic mixture of two enantiomers, R(+)-enantiomer and S(-)-enantiomer.The active ingredient of the test
product formulation is dexrabeprazole, the chirally pure R(+)-enantiomer of rabeprazole. This study aims to compare the pharmacokinetic
properties ofdexrabeprazole containing product (i.e. test product) to racemate rabeprazole containing product (i.e. reference
product) to demonstrate the bioequivalence with respect to the rate and extent of absorption of dexrabeprazolein healthy
male subjects under fasting and fed conditions. Under fasting conditions; for test and reference products, the mean±sd of Cmax
were found 288.374±79.3044 ng/mL and 288.255±89.8262 ng/mL, and the mean±sd of AUC(0-tlast) were found 504.308±211.2707
hr.ng/mL and 572.973±246.9999 hr.ng/mL, respectively. Under fed conditions; for test and reference products, the mean±sd of
Cmax were found 301.094±136.685 ng/mL and 304.202±134.168 ng/mL, and the mean±sd of AUC(0-tlast) were found 576.533±
269.297 hr.ng/mL and 660.652±298.974 hr.ng/mL, respectively. The primary target variables data demonstrate the bioequivalence
of test and reference products with regard to 90% CI for Cmax of 95.23-107.79 and for AUC(0-tlast) of 84.83-90.41 under fasting
conditions and for Cmax of 88.23 – 109.98 and for AUC(0-tlast) of 81.55 – 96.42 under fed conditions. It was shown that the test
product containing dexrabeprazole alone (Rabby-D 10 mg enteric coated tablet, Neutecİlaç San. Tic. A.Ş.-Turkey) and reference
product containing racemate rabeprazole (Pariet® 20 mg gastro-resistant tablet, Eisai Limited European Knowledge Centre-UK)
are bioequivalent in terms of rate and extent of absorption for dexrabeprazole under fasting and fed conditions. Besides, both
products were well tolerated and safe.
2.Introduction
3.Experimental
3.1 Materials
3.2 Methods
3.3 Preparation of Standard and Sample Solutions
4.Results and Discussion
4.1 Chromatographic Conditions Optimization
4.2 Absorption Spectra
4.3 Optimization of Derivatization Reaction and Spectrophotometric Procedure
4.4 Stability of the Drugs - NQS Derivative
4.5 Stoichiometry of Derivatization Reaction
4.6 Validation of the Method
4.1 Application of the Proposed Method to Analysis of AML, HCT and VAL in Tablets
4.8 Reaction Mechanism
5.Conclusion
6.Acknowledgements
7.References
Keywords
Deksrabeprazole; Rabeprazole; Bioequivalence; Gastro-Resistant Tablet; Method Validation.
Introduction
Rabeprazole sodium is a proton pump inhibitör, which is a substituted
benzimidazole known chemically as 2-[[[4-(3methoxypropoxy)-
3-methyl-2-pyridinyl]-methyl]sulfinyl]-1H–benzimidazole sodium
salt. Rabeprazole is a racemic mixture of two enantiomers, R(+)-
enantiomer and S(-)-enantiomer. The active ingredient of the test
product formulation is dexrabeprazole, the chirally pure R(+)-
enantiomer of rabeprazole, which was shown more effective than
the racemate and S(-)-rabeprazole ininhibiting acid-related gastric
lesions in rats [1]. Recent studies have revealed that the pharmacodynamics
of (R)-(+)-rabeprazole were better than those of (S)-
(−)-rabeprazole [1-4].
Absorption of rabeprazole is rapid, with peak plasma levels occurring
approximately 3.5 hours after a 20 mg dose. Peak plasma
concentrations (Cmax) of rabeprazole and AUC are linear over the
dose range of 10 mg to 40 mg. Absolute bioavailability of an oral
20 mg dose (compared to intravenous administration) is about
52% due in large part to pre-systemic metabolism. In healthy
subjects the plasma half-life of rabeprazole is approximately one
hour (range 0.7 to 1.5 hours), and the total body clearance is estimated
to be 283 ± 98 ml/min. Neither food nor the time of day of administration of the treatment affect the absorption of
rabeprazole sodium.
Rabeprazole sodium, as is the case with other members of the
Proton pump inhibitors (PPI class of compounds), is metabolised
through the cytochrome P450 (CYP450) hepatic drug metabolising
system. In vitro studies with human liver microsomes indicated
that rabeprazole sodium is metabolised by isoenzymes of
CYP450 (CYP2C19 and CYP3A4). In these studies, at expected
human plasma concentrations rabeprazole neither induces nor inhibits
CYP3A4. In humans the thioether (M1) and carboxylic acid
(M6) are the main plasma metabolites with the sulphone (M2),
desmethyl-thioether (M4) and mercapturic acid conjugate (M5)
minor metabolites observed at lower levels. Only the desmethyl
metabolite (M3) has a small amount of anti-secretory activity, but
it is not present in plasma. Following a single 20 mg 14C labelled
oral dose of rabeprazole sodium, no unchanged drug was excreted
in the urine. Approximately 90% of the dose was eliminated in
urine mainly as the two metabolites: a mercapturic acid conjugate
(M5) and a carboxylic acid (M6), plus two unknown metabolites.
The remainder of the dose was recovered in faeces [5].
Clinicalbioequivalence studiesarerequired for dexrabeprazole
containing orally administered equivalent products according to
local and globalpharmaceutical regulations [6, 7]. Therefore, two
studies under fasting and fed conditions were conducted to demonstrate
the bioequivalence of the products with respect to the
rate and extent of absorption of deksrabeprazole in healthy male
subjects under fasting and fed conditions.
Subjects and Methods
Ethical Statement
This study was conducted at Farmagen-Good Clinical Practice
Center, Gaziantep, Turkey according to the regulations by Ministry
of Health of the Republic of Turkey which are in compliance
with Declaration of Helsinki and Good Clinical Principles (GCP)
[8]. The protocols and informed consent forms werereviewed and
approved byan independent ethics committee (Erciyes University,
Bioavailability-Bioequivalence Research Ethics Commitee, Kayseri,
Turkey,Approval Date: 17.02.2021for fasting and 09.06.2021
for fed conditions) and Turkish Medicines and MedicalDevices
Agency (Approval Date: 06.04.2021 for fasting and 17.06.2021
for fed conditions). All subjects voluntarily provided signed informed
consent before participation in thestudy.
Study Population and Study Design
All subjects are adult males (aged 18-55 years) with normal weight
according to the a body mass index BMI. The subjects who have
history of drug hypersensitivity (especially to the active and inactive
ingredients of the rabeprazole sodium preparations or
intolerance to any sugar) and who have any history or presence
of clinically significant cardiovascular, renal, hepatic, pulmonary,
metabolic, endocrine, hematological, gastrointestinal, neurological,
psychiatric or other diseases were excluded from the study.
The inclusion and exclusion criteria were established clearly together
with the reasons for withdrawal from the study. The subjects
who were willing to participate in the clinical trial signed the
informed consent form on their own freewill and understood that
they could withdraw from the study anytime without specifying
any reason. Two of the studies were conducted asamonocentric,
open-label, randomised, single oral dose, four-period replicate,
crossover, study in 36 healthy, Caucasian, adult, male, human subjects
under fasting and fed conditions. Both studies consisted of 7
days including 2-days isolation and four consecutive study periods
with a hospitalization of approximately 115 hours and wash-out
between periods “a-day”. Studies were conducted at Farmagen-
Good Clinical Practice Center, Gaziantep, Turkey. The standard
laboratory examinations in blood and urine were done consistent
with the study protocol and the volunteers were checked for presence
of HBsAg, HCV-Ab and HIV-Ab in serum. Also Covid-19
PCR tests were applied to the volunteers before isolation period
and hospitalization. Volunteers were also requested to provide a
urinesample for a drug screen which include “amphetamines, cannabinoids,
benzodiazepines, cocaine, opioids and barbiturates”
and an alcohol breath test before isolation periods. All laboratory
tests were carried out in a certified local laboratory. A total of 36
subjects in each study have been randomised.
Volunteers were isolated for two days at the dorm/hotel; observed
for their well-beings confirming with vital sign measurements;
checking for exclusion criteria and registration of any
concomitant medication. During the isolation period volunteers
consumed the meals served by dorm/hotel administration comply
with the restricted foods and beverages defined in the protocol.
After two night isolation period volunteers were discharged
from dorm/hotel and transferred to the clinic for hospitalization.
A confirmation swab test for COVID-19 before hospitalisation
was assessed. Volunteers were admitted to the clinic with negative
PCR test result.
An evening meal was provided at hospitalization days (total caloric
value of approximately 1200 kcal). On medication days, a
standard lunch (total caloric value is approximately 1200 kcal) was
provided 4 hours after dosing, and a standard evening meal (total
caloric value is approximately 1200 kcal) was provided 10 hours
after dosing in each period. In the study which conducted underfed
conditions a high-fat, high-caloric breakfast (total of approximately
900 to 1000 kcal) was provided between 7:30 and
8:30 a.m. on Day 1 of each period before administration of study
medications.
Investigational Medicinal Products
The test drug used was Rabby-D 10 mg enteric coated tablet (gastroresistant
tablet), Neutecİlaç San. Tic. A.Ş.-Turkey (Batch No:
NI00190-2104 P02; Expiry Date:04.2023); the reference drug
used was Pariet® 20 mg gastro-resistant tablet, Eisai Limited European
Knowledge Centre-UK (Batch No: 127525; Expiry Date:
03.2022).
Blood sampling and Study Assessment
The samples were drawn by a short intravenous catheter at predose
and 1:00, 1:30, 2:00, 2:20, 2:40, 3:00, 3:20, 3:40, 4:00, 4:30,
5:00, 6:00, 7:00, 8:00, 9:00, 10:00, 11:00, 12:00 and 14:00 hours
post-dose in each clinical study period for fasting conditions and
at predose and 1:00, 2:00, 3:00, 3:30, 4:00, 4:30, 5:00, 5:30, 6:00,
6:30, 7:00, 7:30, 8:00, 8:30, 9:00, 10:00, 11:00, 12:00, 14:00 and
16:00 hours post-dose in each clinical study period for fed conditions.
The blood samples (5 ml) were collected into tubes containing K2 EDTA as anti-coagulating agent. After sampling, the
samples were immediately refrigerated at approximately 2-8°C
and will remain there for not more than 20 minutes. Following
the centrifugation (1500 g, 4°C, 10 min), the separated plasma
from each sample weretransferred into two 3.5 mL transparent,
polypropylene tubes. All thealiquoted plasma samples were flash
freezed immediately. The flash frozen samples (aliquoted plasma
samples) were transferred to a deep-freezer and stored at -70°C
until they were transported to the bioanalytical center.
Determination of deksrabeprazole plasma concentrations
The bioanalytical phase of the study has been run at Novagenix
Bioanalytical R&D Center, Ankara, Turkey. In order to avoid bias,
the analytical studies were operated as analytically blinded.
The method used for the determination of dexrabeprazole was
developed and validated by Novagenix Bioanalytical R&D Center,
Ankara in accordance with the earlier published method on stereoselective
pharmacokinetics of rabeprazole [9].
The lower limit of quantification for dexrabeprazole was 1 ng/
mL. Standard curve range for dexrabeprazole was 1 ng/ml to
1200 ng/ml. Dexrabeprazole was extracted from plasma by protein
precipitation using acetonitrile. Finally, the samples were
transferred into a collection plate for analysis.
Materials and Methods
Chemicals and reagents
Reference standard “Dexrabeprazole sodium”was supplied from
Nosch Labs Private Limited, India (Lot no:DRS0050617 with a
retest date of 1st of June 2022) and internal standard (R)-Rabeprazole-
d3 Sodium Salt (Cas no:1216494-11-9, certified purity
95%) was obtained from Toronto Research Chemicals, Canada.
Methanol Acetonitrile ammonium acetate and acetic acid were
purchased from Merck KGaA, Darmstadt, Germany. Ultrapure
(Type 1) water was obtained from Milli-Q plus water purification
system. K2EDTA blank human plasma including haemolysed
and hyperlipidaemic were purchased from Bioivt Elevating
Science,UK and Gaziantep University Farmagen GCP Centre,
Turkey.
Instrument and Conditions
Analyses were performed on a LC-MS/MS system consisting of a
Shimadzu mass spectrometer LCMS-8050 coupled to triple quadrupole
mass spectrometry detector with an electrospray ionization
(ESI) interface and NexeraX2 model LC system (SIL-30AC
autosampler, LC-30AD solvent delivery modules, CTO-10AS vp
column oven, DGU-20A5R degasser unit; Shimadzu Corporation.
Japan). All data were processed by Shimadzu LabSolution
Software version 5.93 (Shimadzu Corporation. Japan).
Chromatographic separations were achieved by using a Daicel
ChiralPAK IC, 4.6 mm I.D. x 150 mmL, 5 μm column and mobile
phase was consisting of acetonitrile and 0.2% acetic acid in
10 mM ammonium acetate solution (80:20, v:v). Flow rate was
0.8 mL/min and the chromatographic run time was 6.5 minutes.
Column temperature was set to 30°C and the autosampler temperature
was 10°C.
The determination of dexrabeprazole is performed in tandem
mass spectrometry operated in the positive ion electrospray ionisation
(ES+).The multiple reaction monitoring (MRM) transitions
were performed at m/z359.90>242.20 for dexrabeprazole
and m/z363.00>245.20 for dexrabeprazole d3.
Preparation of Standard and Quality Control (QC) samples
Stock solutions of (R)-rabeprazole sodium salt were prepared in
methanol:water (1:1) mixture separately for calibration standards
and quality control samples. Final concentrations were 1 mg/mL
and diluted working stock solutions were prepared in methanol.
Internal standard (IS) stock solution was prepared by dissolving
(R)-rabeprazole d3 sodium salt in methanol:water (1:1) mixture
and the final concentration was 0.2 mg/mL stock solution.
Calibration standards were prepared for the concentration levels
of 1, 2, 20, 100, 250, 500, 1080 and 1200 ng/mL and quality control
samples were prepared for the concentration levels of 1, 3,
30, 480 and 960 ng/mL.
Sample Preparation
100 μL plasma was spiked with 50 μL IS working solution (0.7
μg/mL) then protein precipitation was applied with 600 μL acetonitrile.
After centrifugation for 10 minutes at 5500 rpm (4°C), 2
μL upper organic phase was injected to the system.
Method Validation
The method was completely validated according to US-FDA Bioanalytical
Method Validation Guidance [10] and European Medicines
Agency Guideline on Bioanalytical Method Validation [11].
The parameters (selectivity, linearity, lower limit of quantification,
accuracy, precision, dilution integrity, influence of haemolysed
and hyperlipidaemic plasma, drug-drug interaction, carry-over,
recovery, matrix effect,re-injection reproducibility, batch size, stability
of the analyte) were successfully validated.
For selectivity, eight different sources of human blank plasma
(including haemolysed and hyperlipidaemic) were evaluated and
no interference was observed at the retention times and transitions
of dexrabeprazole and dexrabeprazole d3. Eight freshly
prepared calibration standards for dexrabeprazole (1, 2, 20, 100,
250, 500, 1080, 1200 ng/mL) were assayed in each of three validation
batches. For each validation batch, a calibration curve was
acquired by plotting the peak area ratios (peak area analyte/peak
area IS) versus nominal concentration and fitted into the linear
equation using weighing factor 1/C2 as the best fit model for this
curve. The range of precision and accuracy of the back-calculated
concentrations of the calibration curve points were from 0.52%
to 1.84% and from 95.76% to 105.40%, respectively.
The within-batch precision and accuracy were evaluated by analyzing
QC samples at five different concentration levels with six
replicates in a batch.The between-batch precision and accuracy
were determined by analyzing three different batches. The withinbatch
accuracy and precision was 96.70% to 106.94% and 0.45%
to 5.99%, respectively. The between-batch accuracy and precision
was 97.44% to 105.31% and 0.84% to 3.56%, respectively.
Stability evaluation in matrix were processed using freshly prepared
calibration standards and freshly prepared QC samples.
Dexrabeprazole was stable in plasma at room temperature for 5
hours and after 4 freeze thaw cycles. The processed samples were
stable up to 30 hours in autosampler at 10 °C. Dexrabeprazole
was stable in plasma for at least 92 days when stored at -20 ºC
and -70 ºC.
Pharmacokinetic and statistical analyses
In accordance with the bioequivalence recommendation on rabeprazole
sodium delayed-release tablets and the earlier published
assessment reports for rabeprazole sodium containing generic
products, the intra-subject coefficient of variation (ISCV) was
estimated higher than 50% for Cmax and approximately 30% for
AUC(0-tlast). In order to demonstrate bioequivalence with a power
of 80% and a test/reference parameter ratio as 0.95 for a fullyreplicated
crossover design, sample size was estimated as 30. Considering
the possible drop-outs, a sample size of ‘36 volunteers’ was
chosen in a replicate design.
Maximum plasma concentration (Cmax) and area under the curve
from time 0 to the last measurable concentration (AUC(0-tlast)) were
considered as the primary target variables; area under the curve
from time 0 to the infinite time (AUC(0-∞)), time to reach the peak
concentration (tmax), terminal half life (t½), terminal disposition
rate constant (λz) and mean residence time (MRT) were declared
as the secondary target variables in this bioequivalence study.
Cmax and tmax for dexrabeprazole were obtained directly by plasma
concentration-time curves. AUC(0-tlast) was calculated using the
linear-log trapezoidal rule. AUC(0-∞) was calculated by summing
AUC(0-tlast) and extrapolated area. The latter was determined by dividing
the last measuredconcentration by λz which was estimated
byregression of the terminal log-linear plasma concentration time
points.
Databases were automatically processed using the validated program
of Phoenix WinNonlin® version8.3.1.5014 (Certara Inc.,
Pharsight,USA). Obtained PK parameters were processed using
the SAS® software version 9.0.
An analysis of variance (ANOVA) was performed using the General
Linear Model (GLM) procedure, in which sequence, subject
(nested in sequence), period and treatment effects were characterized.
The effects of ANOVA were tested at 5% level of significance.
The 90% confidence intervals were calculated for T/R
ratio of means. The 90% confidence interval of geometric least
square means ratio T/R for AUC(0-tlast) and Cmax for dexrabeprazole
were the primary parameters for the bioequivalence assessment.
In the assessment of bioequivalence, confidence intervals approach
was used. The two one-sided hypothesis at the 5% level of
significance were tested by constructing the 90% confidence intervals
(90% CIs) for the geometric mean ratios of test/reference
products. , In addition, a non-parametric Wilcoxon and median
tests for tmaxwereperformed using SAS procedure NPAR1WAY.
Both databases were automatically processed using the validated
program of Phoenix WinNonlin® version 8.3.1.5014 (Certara
Inc., Pharsight, USA). Obtained PK parameters were processed
using the SAS® software version 9.0
Results
For fasting study, 51 subjects were screened. 36 subjects were randomised
and included into the study. The subjects were divided
into two groups according to the randomisation table. There was
no drop-out and 36 subjects completed the clinical phase of the
study. All of the subjects were Caucasian. The mean±SD age of
subjects was 23.72±6.44 years and the mean±SD body mass index
(BMI) was 24.04±3.15.
For fed study, 47 subjects were screened. 36 subjects were randomised
and included into the study. The subjects were divided
into two groups according to the randomisation table. There was
two drop-outs and 34 subjects completed the clinical phase of the
study. All of the subjects were Caucasian. The mean±SD age of
completed subjects was 26.9±8.50 years and the mean±SD BMI
was 24.7±3.15.
The demographic data of subjects are presented in Table 1. There
was no protocol deviation through the clinical period.
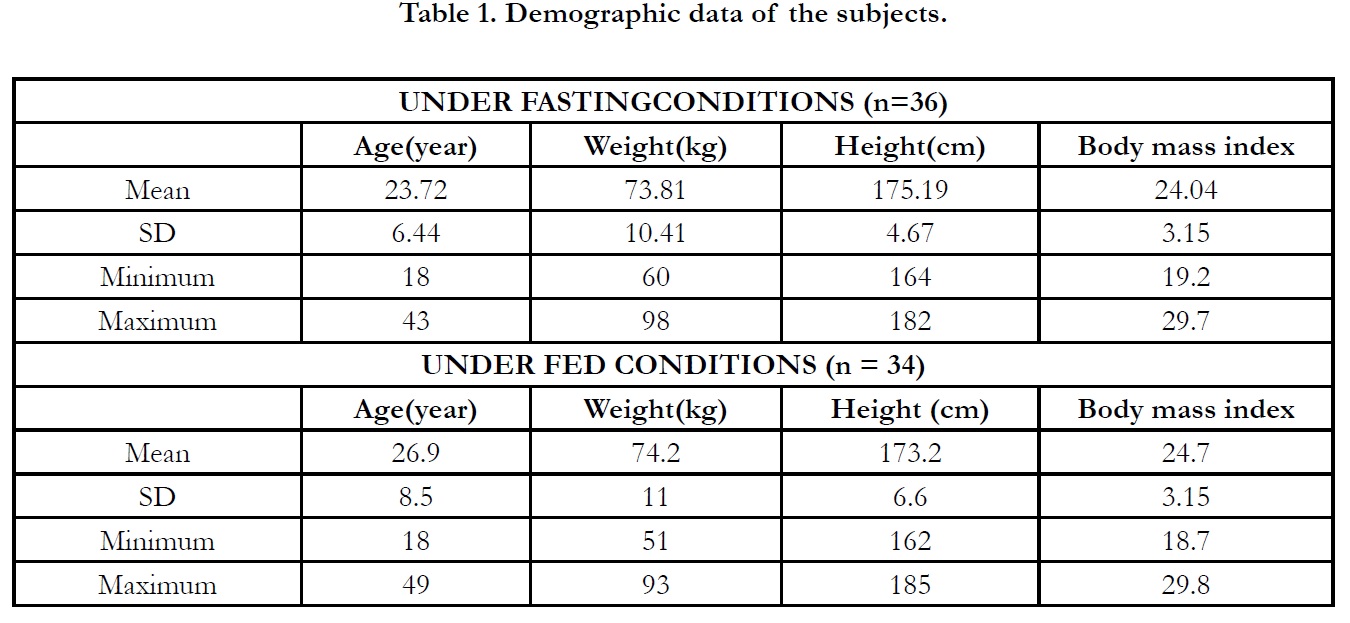

Table 1. Demographic data of the subjects.
Actual time of sampling was used in the estimation of the pharmacokinetic parameters.
In period 2, 3 and 4 at t0.00, the concentrations were found as zero or <LLOQ or less than 5% Cmax for all of the subjects indicating the absence of carry over effect and the washout period of 1 day was sufficient under fasting and fed conditions. The pharmacokinetic parameters for test and reference products are summarised in Table 2 and Table 3 for fasting and fed conditions, respectively. The geometric least square means, ratios and 90% CIs are summarised in Table 4 and Table 5 for fasting and fed conditions, respectively.
Average plasma concentration-time curves and average ln plasma concentration-time curves of test and reference products for single dose of dexrabeprazole under fasting conditions are displayed in Figure 1, respectively.
Average plasma concentration-time curves and average ln plasma concentration-time curves of test and reference products for single dose of dexrabeprazole under fed conditions are displayed in Figure 2, respectively.
Under fasting conditions; for Test and Reference products, the mean±sd of Cmax were found 288.374±79.3044 ng/mL and 288.255±89.8262 ng/mL, and the mean±sd of AUC(0-tlast) were found 504.308±211.2707 hr.ng/mL and 572.973±246.9999 hr.ng/mL, respectively (Table 2).
The ln-transformed geometric least square means ratio (test/ reference, point estimator) for AUC(0-t) was 87.57% and the 90% confidence interval was 84.83% to 90.41%. The ln-transformed geometric least square means ratio (test/reference, point estimator) for Cmax was 101.31% and the 90% confidence interval was 95.23% to 107.79%. Thus, the confidence intervals for AUC(0- t) and Cmax ratios were within the standard acceptance range of 80.00 – 125.00%[4] (Table 4).
For the secondary endpoint data, the median of tmax for Test and Reference product were found 2.333 hr and 3.333 hr, respectively and ranged from 1.00 hr to 6.00 hr and from 2.00 hr to 6.00 hr, respectively. Besides, the mean±sd of t1/2 for Test and Reference product were found 1.910 ± 0.901 hr (ranged from 0.581 hr to 5.100 hr) and 2.181 ± 0.821 hr (ranged from 0.717 hr to 4.424 hr), respectively (Table 2).
Under fed conditions; for Test and Reference products, the mean±sd of Cmax were found 301.094±136.685 ng/mL and 304.202±134.168 ng/mL, and the mean±sd of AUC(0-tlast) were found 576.533± 269.297 hr.ng/mL and 660.652±298.974 hr.ng/ mL, respectively (Table 3).
The ln-transformed geometric least square means ratio (test/ reference, point estimator) for AUC(0-t) was 88.68% and the 90% confidence interval was 81.55% to 96.42% %. The ln-transformed geometric least square means ratio (test/reference, point estimator) for Cmax was 98.50% and the 90% confidence interval was 88.23% to 109.98%. Thus, the confidence intervals for AUC(0- t) and Cmax ratios were within the standard acceptance range of 80.00 – 125.00%. (Table 5).
For the secondary endpoint data, the median of tmax for both Test and Reference product were found 4.000 hr and ranged from 0.00 hr to 16.00 hr. Besides, the mean±sd of t1/2 for Test and Reference product were found 1.907 ± 0.962 hr (ranged from 0.653 hr to 4.945 hr) and 2.120 ± 1.125 hr (ranged from 0.701 hr to 5.829 hr), respectively (Table 3).
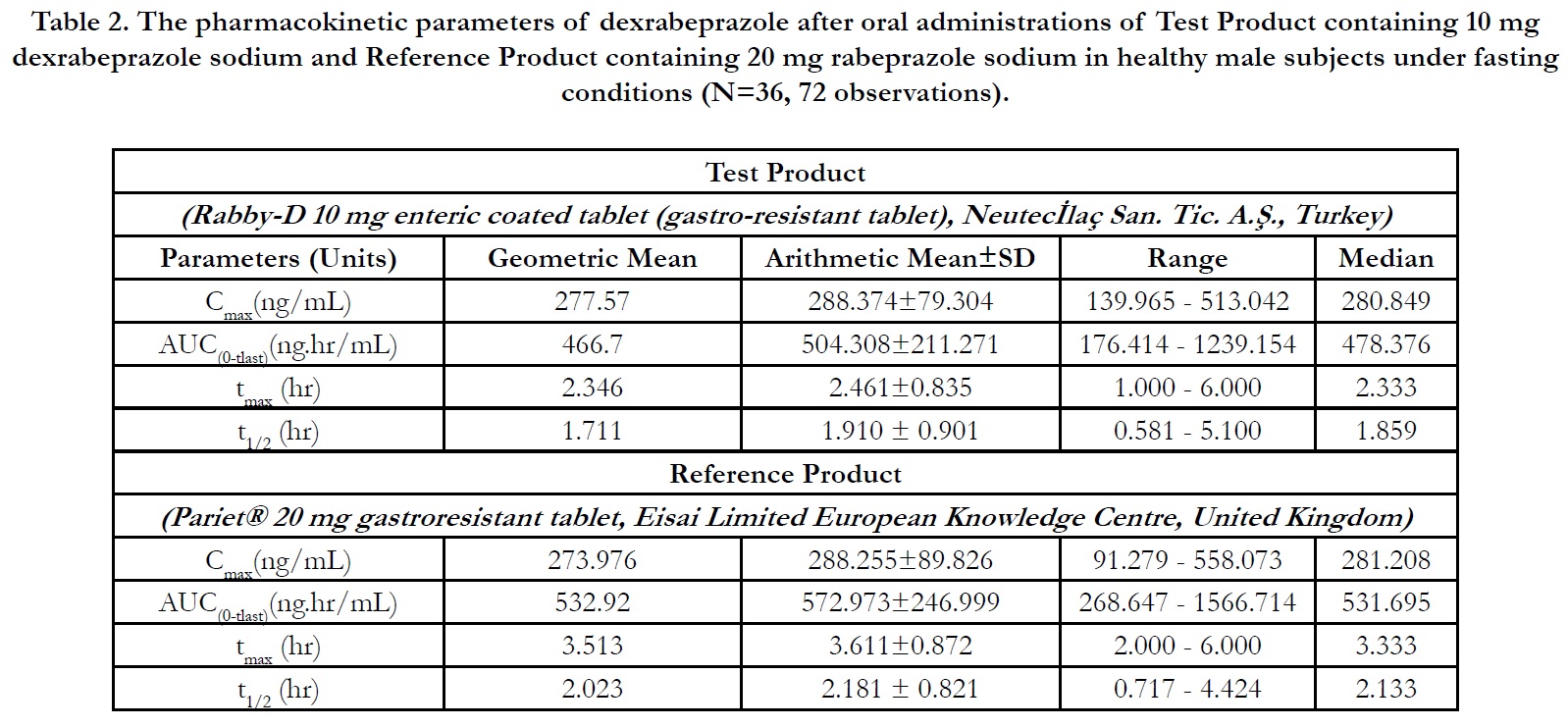
Table 2. The pharmacokinetic parameters of dexrabeprazole after oral administrations of Test Product containing 10 mg dexrabeprazole sodium and Reference Product containing 20 mg rabeprazole sodium in healthy male subjects under fasting conditions (N=36, 72 observations).
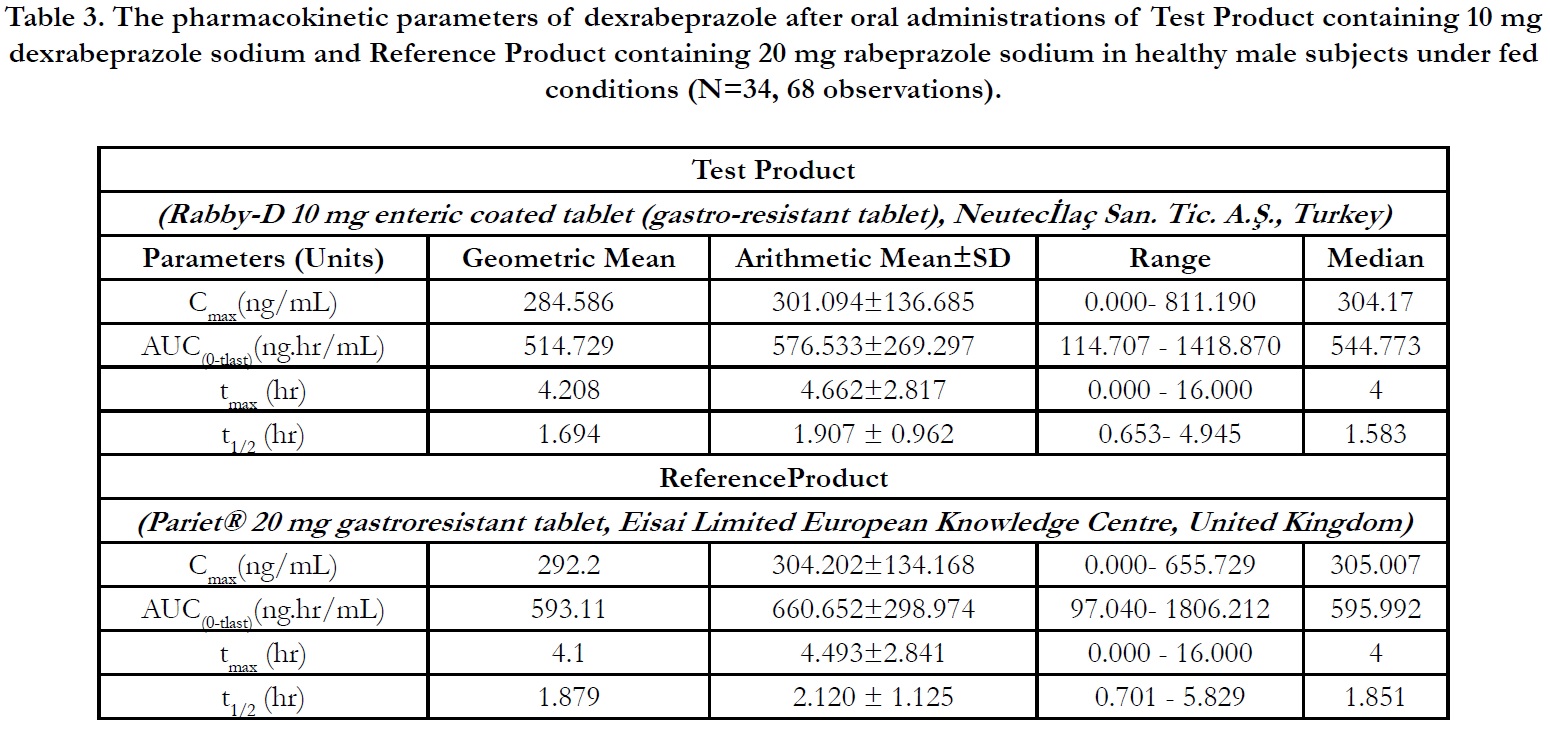
Table 3. The pharmacokinetic parameters of dexrabeprazole after oral administrations of Test Product containing 10 mg dexrabeprazole sodium and Reference Product containing 20 mg rabeprazole sodium in healthy male subjects under fed conditions (N=34, 68 observations).
Statistical Parameters
Tables and Figures 1 & 2
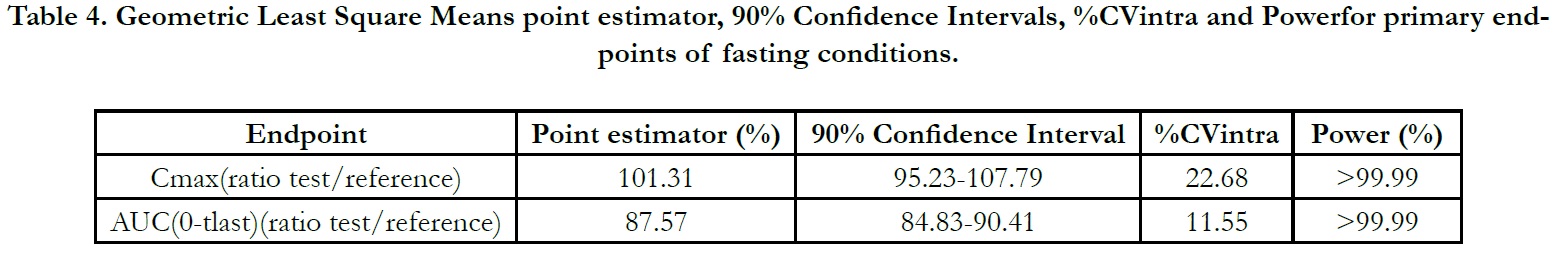
Table 4. Geometric Least Square Means point estimator, 90% Confidence Intervals, %CVintra and Powerfor primary endpoints of fasting conditions.
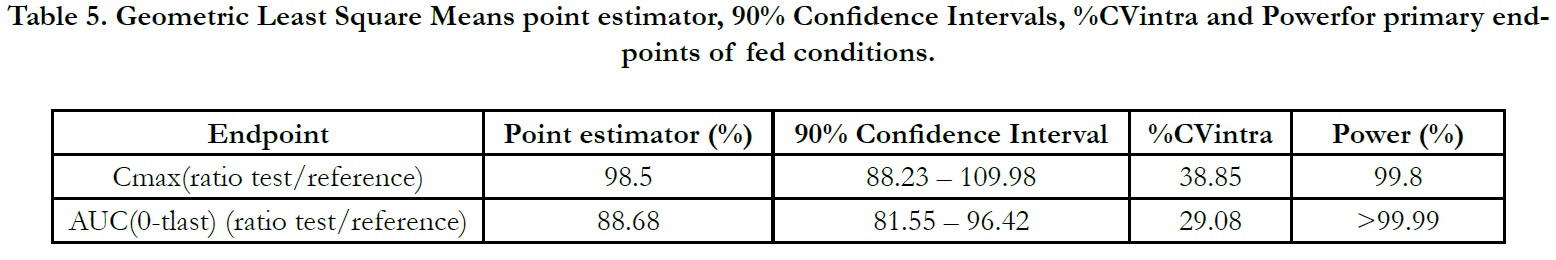
Table 5. Geometric Least Square Means point estimator, 90% Confidence Intervals, %CVintra and Powerfor primary endpoints of fed conditions.
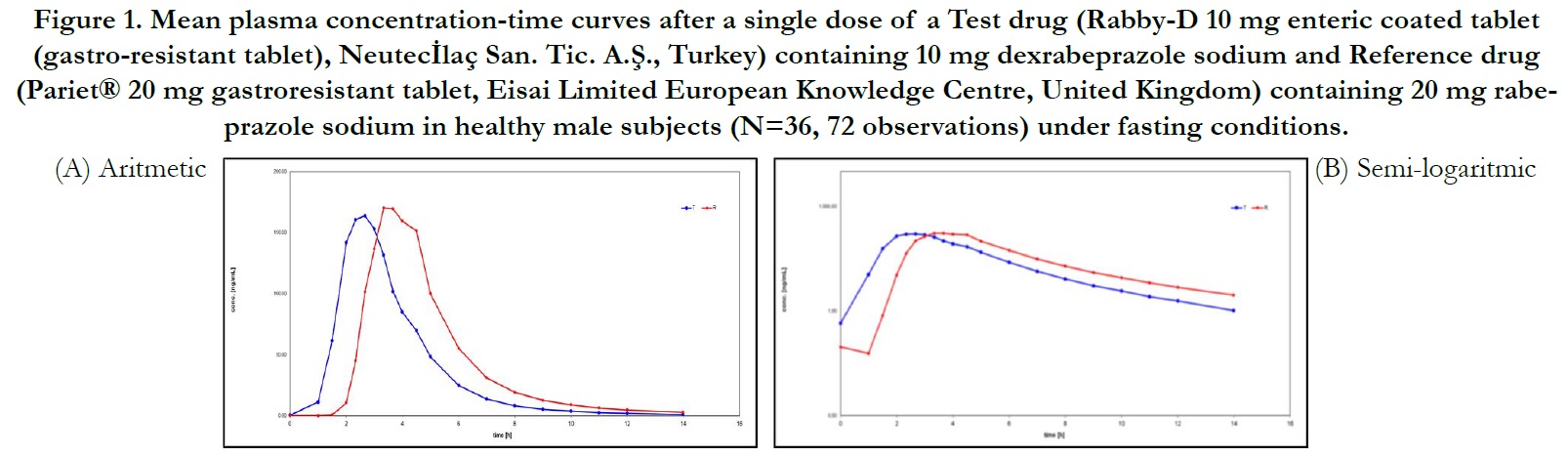

Figure 1. Mean plasma concentration-time curves after a single dose of a Test drug (Rabby-D 10 mg enteric coated tablet (gastro-resistant tablet), Neutecİlaç San. Tic. A.Ş., Turkey) containing 10 mg dexrabeprazole sodium and Reference drug (Pariet® 20 mg gastroresistant tablet, Eisai Limited European Knowledge Centre, United Kingdom) containing 20 mg rabeprazole sodium in healthy male subjects (N=36, 72 observations) under fasting conditions.
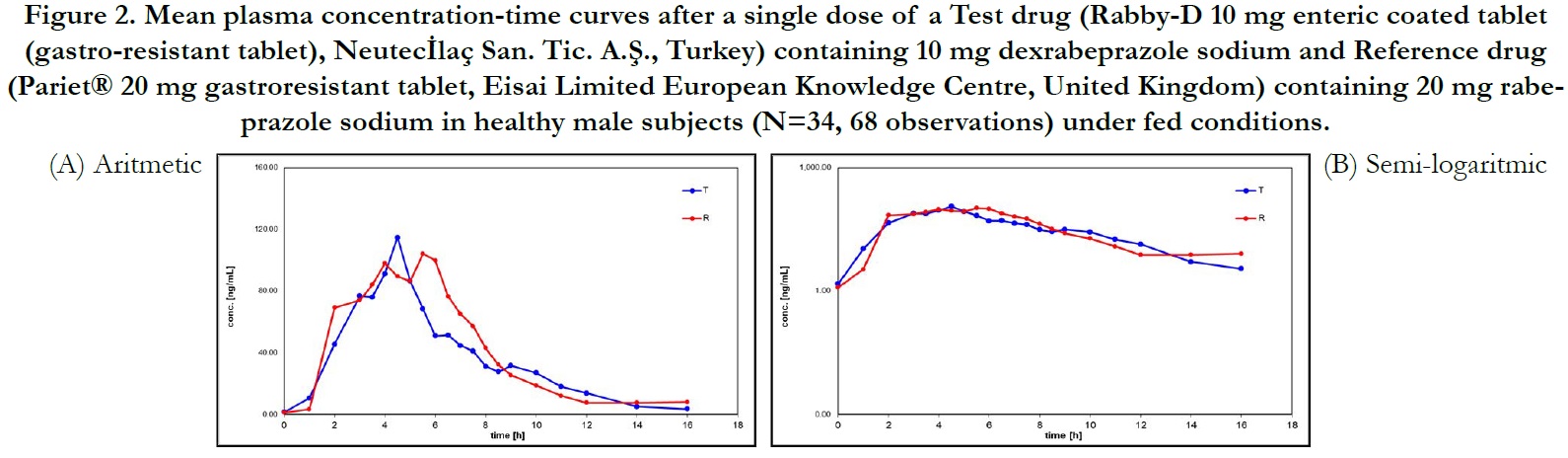

Figure 2. Mean plasma concentration-time curves after a single dose of a Test drug (Rabby-D 10 mg enteric coated tablet (gastro-resistant tablet), Neutecİlaç San. Tic. A.Ş., Turkey) containing 10 mg dexrabeprazole sodium and Reference drug (Pariet® 20 mg gastroresistant tablet, Eisai Limited European Knowledge Centre, United Kingdom) containing 20 mg rabeprazole sodium in healthy male subjects (N=34, 68 observations) under fed conditions.
Discussion
For fasting conditions; the results show that intra-individual variability for Cmax is 22.68%; therefore a wider acceptance range for
Cmax wouldn’t be considered; however 90% confidence limits for
Cmax was found within 80.00% to 125.00%. The conventional acceptance
range (80.00% to 125.00%) for both AUC(0-tlast) and Cmax
are acceptable.
The 90% confidence interval calculated for the primary endpoint,
intra-individual ratios (T/R) for AUC(0-t) of dexrabeprazole was
84.83% to 90.41%. The point estimator was 87.57%. The 90%
confidence interval calculated for the primary endpoint, intraindividual
ratios (T/R) for Cmax of dexrabeprazole was 95.23% to
107.79% with a point estimator of 101.31%. Thus, the confidence
intervals for AUC(0-tlast) and Cmax ratios were within the standard
acceptance range of 80.00 – 125.00% under fasting conditions.
For fed conditions; bioequivalence evaluation was based on the
geometric LSM ratios T/R of the primary endpoint parameters,
AUC(0-tlast) and Cmax of dexrabeprazole. The 90% confidence interval
calculated for the primary endpoint, intra-individual ratios
(T/R) for AUC(0-tlast) of dexrabeprazole was 81.55% to 96.42 and
the point estimator was 88.68%. The 90% confidence interval calculated
for the primary endpoint, intra-individual ratios (T/R) for
Cmax of dexrabeprazole was 88.23% to 109.98% with a point estimator
of 98.50%. The intra-individual variability for Cmax under
fed conditions was 38.85%; therefore a wider acceptance range
for Cmax would be acceptable. However, as the 90% confidence intervals
for both primary parameters were found within the standard
bioequivalence acceptance range of 80.00% to 125.00%, a
wider acceptance range was not required to be applied. Thus the
confidence intervals for AUC(0-tlast) and Cmax ratios were within the
standard acceptance range of 80.00 – 125.00% under fed conditions.
tmaxswere analyzed using Wilcoxon test. There was a significant
difference between two formulations with a significance level of
5% (p<0.0001) under fasting conditions and there was no significant
difference between two formulations with a significance level
of 5% (p>0.05) under fed conditions.
Conclusions
According to the Study Protocols, the AUC(0-t) and Cmax parameters
for dexrabeprazole were used to assess bioequivalence. The
results confirm that the 90% confidence intervals for Test to
Reference ratios of the geometric least squares means for AUC(0-
t) and Cmax were within the bioequivalence acceptance range of
80.00 to 125.00% under fasting and fed conditions.
The test product containing dexrabeprazole (Rabby-D 10 mg enteric
coated tablet, Neutecİlaç San. Tic. A.Ş.-Turkey) and reference
product containing rabeprazole (Pariet® 20 mg gastro-resistant
tablet, Eisai Limited European Knowledge Centre-UK)are bioequivalent
in terms of rate and extent of absorption for dexrabeprazoleunder fasting and fed conditions. The two bioequivalence
studies demonstrated equivalent system exposure of dexrabeprazole
following oral dose given as dexrabeprazole alone (as test
product Rabby-D 10 mg enteric coated tablet, Neutecİlaç San.
Tic. A.Ş.-Turkey) and as racemate rabeprazole (referenceproduct,
Pariet® 20 mg gastro-resistant tablet, Eisai Limited European
Knowledge Centre-UK). The test and reference formulations
demonstrated similar tolerability. No adverse event and serious
adverse event was registered during the studies. Both study drugs
were well-tolerated and considered to be safe.
Acknowledgements
This study was funded by Genera Pharma AG., Wollerau, Switzerland.
Clinical part of this study was conducted at Farmagen Good
Clinical Practice Center, Gaziantep, Turkey, the bioanalyticanalysis
were carried out by Novagenix Bioanalytical Drugs R&D,
Ankara, Turkey and the biostatistical analysis were performed at
TNC İlaçAraştırmaGeliştirmeDanışmanlık San. ve Tic. Ltd. Şti.,
İstanbul, Turkey.
Data Availability
The data that support the findings of this study are available from
the corresponding author upon reasonable request. Some data
may not be made available because of privacy or ethical restrictions.
References
- Bodhankar SL, Jain BB, Ahire BP, Daude RB, Shitole PP.The effect of rabeprazole and its isomers on aspirin and histamineinduced ulcers in rats. Indian J Pharmacol 2006;38:357-358.
- Pai V, Pai N. Randomized, double-blind, comparative study of dexrabeprazole 10 mg versus rabeprazole 20 mg in the treatment of gastroesophageal reflux disease. World J Gastroenterol. 2007 Aug 14;13(30):4100-2. PubMed PMID: 17696229.
- Miura M, Satoh S, Tada H, Habuchi T, Suzuki T. Stereoselective metabolism of rabeprazole-thioether to rabeprazole by human liver microsomes. Eur J ClinPharmacol. 2006 Feb;62(2):113-7. PubMed PMID: 16389533.
- Miura M, Kagaya H, Tada H, Uno T, Yasui-Furukori N, Tateishi T, et al. Enantioselective disposition of rabeprazole in relation to CYP2C19 genotypes. Br J ClinPharmacol. 2006 Mar;61(3):315-20. PubMed PMID: 16487225.
- SPC, Electronic Medicines Compendium, PARIET 10 mg & 20 mg gastroresistant tablet, Eisai Ltd, Last revised on 11/10/2017.
- Guideline on The Investigation of Bioequivalence. CPMP/EWP/ QWP/1401/98 Rev.1/Corr., London, EMA; 2010.
- Guidance for Industry. Bioavailability and bioequivalence studies for orally administered drug products- General Considerations.FDA, CDER; 2014.
- The Guidance for GCP, published by the Ministry of Health of Turkey. Circular, 13.11.2015.
- Sun LN, Shen YW, Ying YW, Li D, Li TF, Zhang XH, et al. Stereoselective pharmacokinetics of (R)-(+)- and (S)-(-)-rabeprazole in human using chiral LC-MS/MS after administration of rabeprazole sodium enteric-coated tablet. Chirality. 2018 Dec;30(12):1277-1286. PubMed PMID: 30321480.
- Guidance for Industry. Bioanalytical Method Validation.FDA, CDER; 2018.
- Guideline on Bioanalytical Method Validation, EMEA/CHMP/ EWP/192217/2009 0Rev.1 Corr.2, London; 2011.